Could It Be CF?
CF Signs & Symptoms to Look Out For
Sinus and lower respiratory symptoms1

- Allergic bronchopulmonary aspergillosis
- Nontuberculous mycobacterial (NTM) infection
GI, reproductive, other symptoms1

- Abnormal GI motility, rectal prolapse
- Elevated liver enzymes, cirrhosis
- Fat-soluble vitamin deficienciesa
NOTE: Common symptoms at first presentation of CF are in bold type; uncommon are in regular type.
Have you considered CF?
These are some of the clinical signs and symptoms that are consistent with CF
Have you considered CF?
These are some of the clinical signs and symptoms that are consistent with CF
Have you considered CF?
These are some of the clinical signs and symptoms that are consistent with CF
What is CF?
Cystic fibrosis (CF) is a systemic, multi-organ disease that is caused by mutations in both cystic fibrosis transmembrane conductance regulator (CFTR) alleles.2 Mutations in the CFTR gene may cause impaired cellular processing and trafficking, including an absence of or reduction in channel numbers, channel function, or both.2 Some presenting manifestations of CF can include chronic sinusitis, bronchiectasis, NTM infection, recurrent pancreatitis, respiratory symptoms and/or male infertility.1
Reduced CFTR quantity


Reduced CFTR function

Impact on the lungs
The insufficient flow of chloride and water create mucus buildup that can result in abnormal mucociliary clearance and airway inflammation. This leads to progressive lung damage, including mucus plugging, air trapping, air wall thickening, and bronchiectasis.2-4
Bronchiectasis is a common symptom of CF5
Bronchiectasis is a common symptom of CF5
Bronchiectasis is a common
symptom of CF5
Rates of CF testing in bronchiectasis populations have been estimated to be between 5.5% and 12%.6,7b
Other symptoms consistent with CFTR dysfunction8,9

Upper respiratory symptoms
Rhinosinusitis
Lower respiratory symptoms
NMT infection or isolation of Pseudomonas aeruginosa or Staphylococcus aureus
Other
Extrapulmonary features
- Malabsorption/GI symptoms
- Pancreatitis
- Intestinal obstruction
Male infertility
CF is one possible cause of bronchiectasis
aMay present as ecchymosis, anemia, edema, night blindness, or skin rash.1
bStudies of US rates are not published; these percentages are based on testing rates in Italy and the United Kingdom, respectively.6,7
Reasons for Late Diagnosis10-13

Born before implementation of or in a country without NBSa

Racial and ethnic disparities in CFTR mutations contained in standard NBS panels

Harbor rare CFTR mutations not included in standard genetic panels

Misdiagnosed for another pulmonary disease (e.g., bronchiectasis, NTM infection)

Lack of access to healthcare services
Delayed CF diagnosis may ultimately lead to disparities in treatment, care, and outcomes over time14
Delayed CF diagnosis may ultimately lead to disparities in treatment, care, and outcomes over time14
Delayed CF diagnosis may ultimately lead to disparities in treatment, care, and outcomes over time14
aWhile all states now perform NBS for CF, there are variations in the approach and some approaches may miss some CF diagnoses.13
CF Can Affect a Diverse Population
CF has traditionally been thought of as a disease affecting exclusively people of European descent.15 However, increasing numbers of people with CF of non-White backgrounds have been diagnosed and reported in the US CFFPR.16
Diversity in age distribution of CF diagnosis
Diversity in age distribution of CF diagnosis
Diversity in age distribution
of CF diagnosis
Although universal CF NBS was implemented in the United States in 2009 and enabled early identification of CF, there are people with CF who are not diagnosed until adulthood.13a,16


CF is caused by numerous mutations in the CFTR gene that vary across racial and ethnic groups14,18,19
- The most common CFTR mutation is F508del. 85.4% of those in the US CFFPR who have been genotyped have at least 1 copy of this variant16
- Prevalence of F508del mutation varies by ancestry18
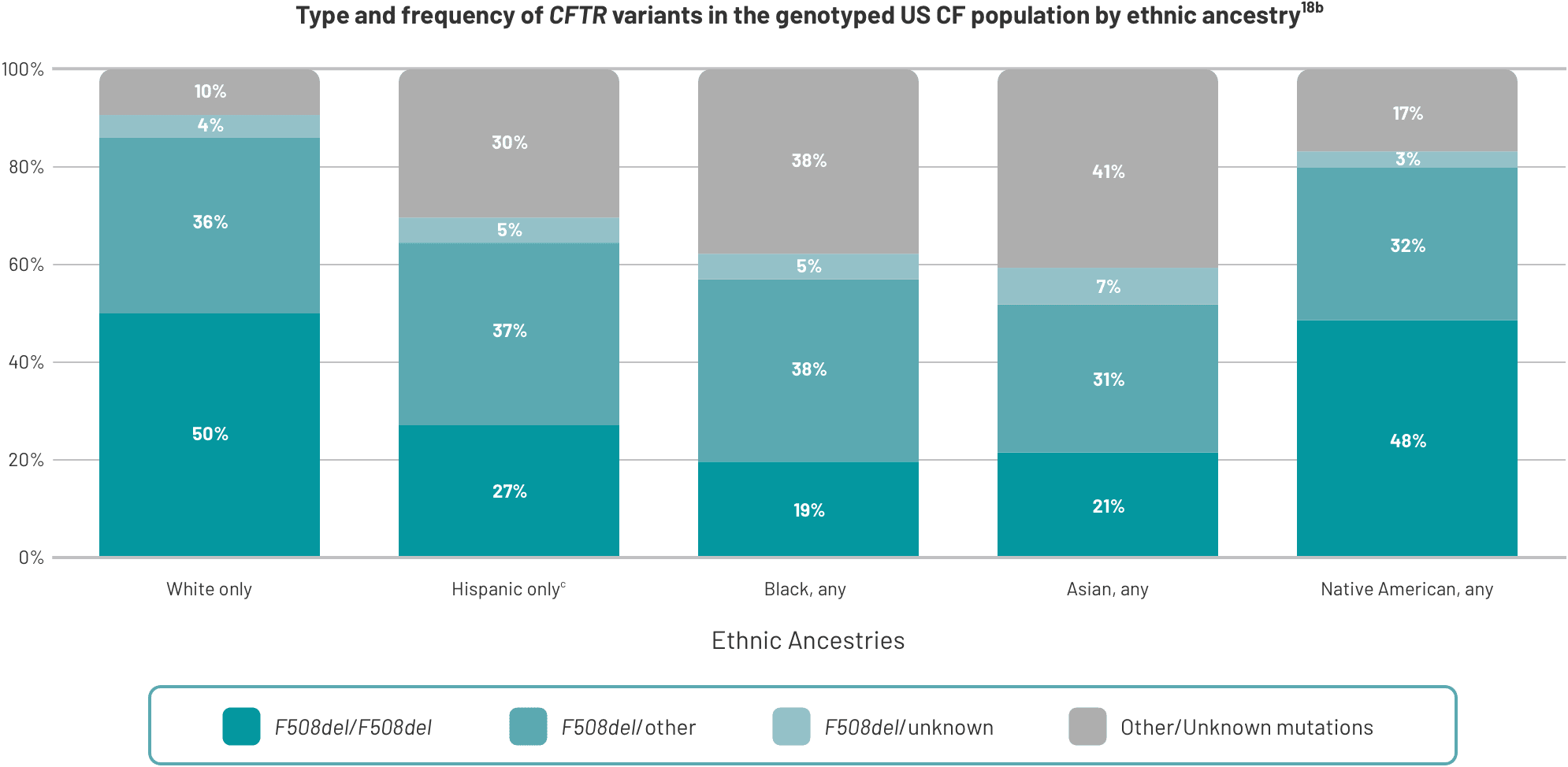

Hispanic, Black, Asian, and Native American people are more likely to have rare CFTR mutations18
Hispanic, Black, Asian, and Native American people are more likely to have rare CFTR mutations18
Hispanic, Black, Asian, and Native American people are more likely to have rare CFTR mutations18
aData from the 2022 US CFFPR.
bThis study was in patients diagnosed with CF from the US CFFPR.18
cPercentages are rounded and may not add up to exactly 100%.
Considerations to Investigate Whether It's CF
Common diagnostic tests for CF
- Sweat chloride test: Sweat chloride concentration is the standard for diagnosing CF and an important marker of CFTR function20a
- CFTR genotyping: Genotyping is potentially helpful in the diagnostic workup for CF20
Consider whether it is CF by…

IDENTIFYING
Older, non-White patients with bronchiectasis or other symptoms suggestive of CFTR dysfunction1,5,16

REFERRING
Appropriate individuals for sweat chloride testing, regardless of race/ethnicity or age1,14,16,17,20

SENDING
Appropriate individuals to a CF care center for additional diagnostic workups and/or care
Click here to find a CF care center
Benefits of diagnosis and treatment
- Earlier and accurate diagnosis of CF in all patients is important due to clinical and psychological consequences21
- Treatment is available for people with CF in the U.S. through specialized CF care centers22,23
- Once diagnosed, treatments are available for patients with CF that can contribute to improve quality of life and life expectancy14,16
- The median age of survival in people with CF has increased in the last 10-20 years due to earlier diagnosis and treatment10,16
- The median predicted survival age of an individual born with CF in 2022 was 68.2 years (95% CI: 63.0, 76.2 years)16b
The treatment landscape continues to expand for those with rarer CF mutations and for the plurality of undiagnosed populations
The treatment landscape continues to expand for those with rarer CF mutations and for the plurality of undiagnosed populations
The treatment landscape continues to expand for those with rarer CF mutations and for the plurality of undiagnosed populations
aWhen accompanied by clinical suspicion, such as positive newborn screen, clinical features, and/or family history of CF.
bThis prediction does not consider the potential impact of CFTR modulators on younger individuals with CF or additional future improvements in clinical care.16
CFFPR, Cystic Fibrosis Foundation Patient Registry; CI: confidence interval; GI, gastrointestinal; NBS, newborn screening; NTM, nontuberculous mycobacterial
You May Also Be Interested In


These organizations are separate from and not associated with Vertex Pharmaceuticals Incorporated.
References: 1. Sosnay PR, White TB, Farrell PM, et al. Diagnosis of cystic fibrosis in nonscreened populations. J Pediatr. 2017;181(suppl):S52-S57.e2. doi:10.1016/j.jpeds.2016.09.068. 2. Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A. Cystic fibrosis. Nat Rev Dis Primers. 2015;1:15010. doi:10.1038/nrdp.2015.10. 3. Morrison CB, Markovetz MR, Ehre C. Mucus, mucins, and cystic fibrosis. Pediatr Pulmonol. 2019;54(suppl 3):S84-S96. doi:10.1002/ppul.24530. 4. Wielpütz MO, Puderbach M, Kopp-Schneider A, et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med. 2014;189(8):956-965. doi:10.1164/rccm.201309-1659OC. 5. Sly PD, Gangell CL, Chen L, et al; AREST CF Investigators. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368(21):1963-1970. doi:10.1056/NEJMoa1301725. 6. Hill AT, Routh C, Welham S. National BTS bronchiectasis audit 2012: Is the quality standard being adhered to in adult secondary care? Thorax. 2014;69(3):292-294. doi:10.1136/thoraxjnl-2013-203739. 7. Aliberti S, Hill AT, Mantero M, et al; SIP Bronchiectasis Audit Working Group. Quality standards for the management of bronchiectasis in Italy: a national audit. Eur Respir J. 2016;48(1):244-248. doi:10.1183/13993003.00232-2016. 8. Redondo M, Keyt H, Dhar R, Chalmers JD. Global impact of bronchiectasis and cystic fibrosis. Breathe (Sheff). 2016;12(3):222-235. doi:10.1183/20734735.007516. 9. Flume PA, Chalmers JD, Olivier KN. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet. 2018;392(10150):880-890. doi:10.1016/S0140-6736(18)31767-7. 10. McBennett KA, Davis PB, Konstan MW. Increasing life expectancy in cystic fibrosis: advances and challenges. Pediatr Pulmonol. 2022;57(suppl 1):S5-S12. doi:10.1002/ppul.25733 11. Cystic Fibrosis Trust. Cystic fibrosis: late diagnosis. Factsheet. August 2020. Accessed September 9, 2024. https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/Late%20diagnosis%20factsheet%20Sep%202020.pdf. 12. McColley SA, Martiniano SL, Ren CL, et al. Disparities in first evaluation of infants with cystic fibrosis since implementation of newborn screening. J Cyst Fibros. 2023;22(1):89-97. 13. Sontag MK, Miller JI, McKasson S, et al. Newborn screening for cystic fibrosis: a qualitative study of successes and challenges from universal screening in the United States. Int J Neonatal Screen. 2022;8(3):38. doi:10.3390/ijns8030038. 14. Cystic Fibrosis Foundation. Dispelling misconceptions about CF. Accessed September 9, 2024. 15. Guo J, Garratt A, Hill A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J Cyst Fibros. 2022;21(3):456-462. doi:10.1016/j.jcf.2022.01.009. 16. Cystic Fibrosis Foundation Patient Registry. 2022 Annual Data Report to the Center Directors. Bethesda, Maryland. ©2022 Cystic Fibrosis Foundation. Accessed September 9, 2024. https://www.cff.org/media/31216/download. 17. 2022 Cystic Fibrosis Foundation Patient Registry Highlights. Bethesda, Maryland. ©2023 Cystic Fibrosis Foundation. Accessed September 9, 2024. https://www.cff.org/media/29691/download. 18. Schrijver I, Pique L, Graham S, Pearl M, Cherry A, Kharrazi M. The spectrum of CFTR variants in nonwhite cystic fibrosis patients: implications for molecular diagnostic testing. J Mol Diagn. 2016;18(1):39-50. doi:10.1016/j.jmoldx.2015.07.005. 19. CFTR2. Clinical and functional translation of CFTR2 Variant List. Last updated on April 7, 2023. Accessed September 9, 2024. https://cftr2.org/mutations_history. 20. Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181(suppl):S4-S15.e1. doi:10.1016/j.jpeds.2016.09.064. 21. Southern KW, Addy C, Bell SC, et al. Standards for the care of people with cystic fibrosis; establishing and maintaining health. J Cyst Fibros. 2024;23(1):12-28. doi:10.1016/j.jcf.2023.12.002 22. Cystic Fibrosis Foundation. Care centers. Accessed September 9, 2024. https://www.cff.org/managing-cf/care-centers 23. Cystic Fibrosis Foundation. Your CF care team. Accessed September 9, 2024. https://www.cff.org/managing-cf/your-cf-care-team